Publication
Dominik's paper accepted at Journal of Theoretical Biology
D. Schrempf, B.Q. Minh, N. De Maio, A. von Haeseler, and C. Kosiol (2016) Reversible polymorphism-aware phylogenetic models and their application to tree inference. J. Theor. Biol., in press. http://dx.doi.org/10.1016/j.jtbi.2016.07.042
The associated IQ-TREE PoMo version and user guide are available at:
http://www.iqtree.org/#variant
Highlights
- Species tree inference from genome-wide population data.
- Takes incomplete lineage sorting into account.
- Analytical solution of stationary distribution and formal proof of reversibility.
- Reversibility ensures swiftness and stability.
- Increase of sample size per species improves estimations without raising runtime.
- Comparison to the Wright-Fisher diffusion.
Abstract: We present a reversible Polymorphism-Aware Phylogenetic Model (revPoMo) for species tree estimation from genome-wide data. revPoMo enables the reconstruction of large scale species trees for many within-species samples. It expands the alphabet of DNA substitution models to include polymorphic states, thereby, naturally accounting for incomplete lineage sorting. We implemented revPoMo in the maximum likelihood software IQ-TREE. A simulation study and an application to great apes data show that the runtimes of our approach and standard substitution models are comparable but that revPoMo has much better accuracy in estimating trees, divergence times and mutation rates. The advantage of revPoMo is that an increase of sample size per species improves estimations but does not increase runtime. Therefore, revPoMo is a valuable tool with several applications, from speciation dating to species tree reconstruction.
Olga's paper accepted at Systematic Biology
O. Chernomor, A. von Haeseler, and B.Q. Minh (2016) Terrace Aware Data Structure for Phylogenomic Inference from Supermatrices. Syst. Biol., 65, in press.
http://dx.doi.org/10.1093/sysbio/syw037
Abstract: In phylogenomics the analysis of concatenated gene alignments, the so-called supermatrix, is commonly accompanied by the assumption of partition models. Under such models each gene, or more generally partition, is allowed to evolve under its own evolutionary model. Although partition models provide a more comprehensive analysis of supermatrices, missing data may hamper the tree search algorithms due to the existence of phylogenetic (partial) terraces. Here, we introduce the phylogenetic terrace aware (PTA) data structure for the efficient analysis under partition models. In the presence of missing data PTA exploits (partial) terraces and induced partition trees to save computation time. We show that an implementation of PTA in IQ-TREE leads to a substantial speedup of up to 4.5 and 8 times compared with the standard IQ-TREE and RAxML implementations, respectively. PTA is generally applicable to all types of partition models and common topological rearrangements thus can be employed by all phylogenomic inference software.
Jana's paper accepted at Nucleic Acids Research
J. Trifinopoulos, L.-T. Nguyen, A. von Haeseler, and B.Q. Minh (2016) W-IQ-TREE: a fast online phylogenetic tool for maximum likelihood analysis. Nucleic Acids Res., 44, W232-W235. http://dx.doi.org/10.1093/nar/gkw256
The associated IQ-TREE web server is available at:
http://iqtree.cibiv.univie.ac.at
Abstract: This article presents W-IQ-TREE, an intuitive and user-friendly web interface and server for IQ-TREE, an efficient phylogenetic software for maximum likelihood analysis. W-IQ-TREE supports multiple sequence types (DNA, protein, codon, binary and morphology) in common alignment formats and a wide range of evolutionary models including mixture and partition models. W-IQ-TREE performs fast model selection, partition scheme finding, efficient tree reconstruction, ultrafast bootstrapping, branch tests, and tree topology tests. All computations are conducted on a dedicated computer cluster and the users receive the results via URL or email. W-IQ-TREE is available at http://iqtree.cibiv.univie.ac.at. It is free and open to all users and there is no login requirement.
Olga's paper accepted at Journal of Computational Biology
Congrats to Olga for an ultrafast acceptance of her paper (Chernomor et al., 2015) at Journal of Computational Biology. It was accepted just after 8 days of submission!
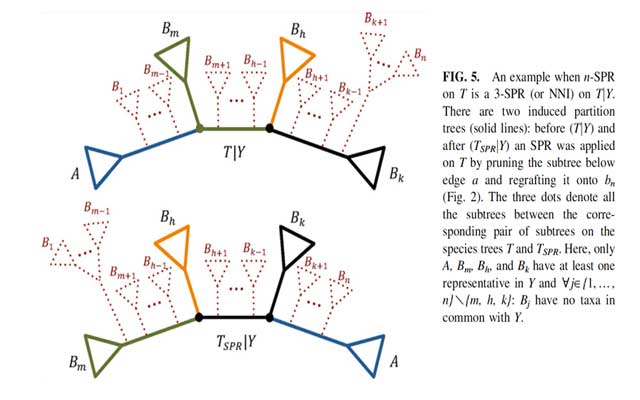
Abstract: In phylogenomic analysis the collection of trees with identical score (maximum likelihood or parsimony score) may hamper tree search algorithms. Such collections are coined phylogenetic terraces. For sparse supermatrices with a lot of missing data, the number of terraces and the number of trees on the terraces can be very large. If terraces are not taken into account, a lot of computation time might be unnecessarily spent to evaluate many trees that in fact have identical score. To save computation time during the tree search, it is worthwhile to quickly identify such cases. The score of a species tree is the sum of scores for all the so-called induced partition trees. Therefore, if the topological rearrangement applied to a species tree does not change the induced partition trees, the score of these partition trees is unchanged. Here, we provide the conditions under which the three most widely used topological rearrangements (nearest neighbor interchange, subtree pruning and regrafting, and tree bisection and reconnection) change the topologies of induced partition trees. During the tree search, these conditions allow us to quickly identify whether we can save computation time on the evaluation of newly encountered trees. We also introduce the concept of partial terraces and demonstrate that they occur more frequently than the original “full” terrace. Hence, partial terrace is the more important factor of timesaving compared to full terrace. Therefore, taking into account the above conditions and the partial terrace concept will help to speed up the tree search in phylogenomic inference.”
Further reading:
O. Chernomor, B.Q. Minh, and A. von Haeseler (2015) Consequences of Common Topological Rearrangements for Partition Trees in Phylogenomic Inference. J. Comput. Biol., in press. (DOI: 10.1089/cmb.2015.0146, PMID: 26448206)
Announcement
Parallel IQ-TREE MPI pre-release
We would like to announce a new MPI parallel version of IQ-TREE for use in computer cluster. In addition to the existing OpenMP parallelization that distributes the tree likelihood computation among CPU cores, IQ-TREE now can employ MPI to distribute the tree search among different computing nodes.
The MPI version of IQ-TREE provides better parallel efficiency than OpenMP and can also be used within a multi-core PC (only Linux and Mac OS X are supported). Note that the memory requirement of the MPI version is proportional to the number of processes. Thus, to avoid excessive memory consumption one can also combine OpenMP and MPI in IQ-TREE (this is called hybrid parallelization).
Information about how to compile and run the program can be found in our documentation page.
IQ-TREE Heterotachy pre-release
We have released an IQ-TREE variant which implements a heterotachy mixture model accounting for heterotachous evolution — rate heterogeneity among sites and lineages. This version can be downloaded from here:
http://www.iqtree.org/#variant
A user guide is available at http://www.iqtree.org/doc/Heterotachy-Models/.
Please note that this is still a pre-release version — so suggestions and feedback are welcome!
New IQ-TREE web site
We are happy to announce the launch of the new IQ-TREE web site immediately available at the same URL:
http://www.cibiv.at/software/iqtree
Notably we updated extensive online user documentation for using IQ-TREE including quick start guide, tutorials, command reference, maximum likelihood models and compilation guide. This is to replace the outdated pdf manual.
The IQ-TREE source code is now hosted at github:
https://github.com/Cibiv/IQ-TREE
for developers’ convenience.
Phd dissertation
Tung's PhD defense
Congrats to Lam-Tung Nguyen! He just successfully defended his PhD dissertation today entitled:
Computational methods for fast and accurate phylogenetic inference
Olga's PhD defense
Congrats to Olga Chernomor! She just successfully defended her PhD dissertation today entitled:
Phylogenomics: theory, algorithms and applications
Also available at university library: http://othes.univie.ac.at/39935/
Opinion
IQTree vs RAxML for phylogenetic tree construction
A user opinion on IQ-TREE:
The results look promising with tree topology, branch length, and support values showing broad correlation (see below for data). The fact that this is an actively developed piece of software, with good documentation and some good peer reviewed papers gives me confidence to try this for the next few phylogenetic analyses I need to run.
Read the original article here:
http://bitsandbugs.org/2016/01/25/iqtree-vs-raxml-for-phylogenetic-tree-construction/